
Sooner or later everybody has to pass a urine drug test. Not everybody has clean urine, however.
Failing a test for any kind of drug can cost you a job, a promotion, college entrance or even put you behind bars.
If somebody uses drugs, the solution of how to pass a drug test is quite clear: almost everybody thinks of using synthetic urine.
But does synthetic urine even work for lab tests?
The best synthetic urine brands are still extremely reliable. Here’s what the result of using real ‘dirty’ urine vs. a 2022 upgraded synthetic urine looks like:
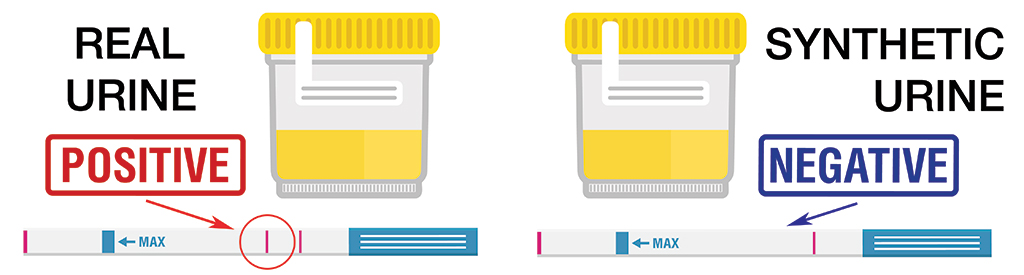
Given how many people use synthetic urine to pass a urine drug test, we looked into what synthetic urine actually is, what it is used for and which synthetic urine brands offer the most reliable fake pee.
Note: Nobody feels 100% at ease when using synthetic urine. However, you just happen to be put in a situation where this is the only way you will have a chance (and an excellent chance at that) to pass a drug test with flying colors. That’s why having the best synthetic urine does ease your mind significantly.
Best Synthetic Urine Brands In 2022
Reliability is the single most important factor when it comes to synthetic urine kits. We are, after all, buying the surest way to pass a drug test. The reliability of that kit is of paramount importance.
The most reliable synthetic urine brands are the ones with a good track record and, even more importantly, with the synthetic urine formulas upgraded to 2022 drug-testing standards. Here is the full list of the best synthetic urine kits currently on the market:
| Synthetic Urine: | #1 Sub Solution | #2 Quick Luck | #3 Quick Fix 6.2 | #4 Powdered Urine |
|---|---|---|---|---|
| Photo: |  |  |  | |
| Track-Record: |  | |  |  |
| Reliability: | | | |  |
| Quality: |  | | | |
| Price: | $$$$ | $$$$ | $$$$ | $$$$ |
| Avg. Review: |  |  |  |  |
| Availability: | Check Here | Check Here | Check Here | Check Here |
According to this formula, it’s clear to see which fake pee kits are the best to use with current laboratory testing standards:
#1 Clear Choice Sub Solution: Best Kit For The Past 5 Years
In the world of fake pee manufacturing, a lot of fake pee is, well, actually fake. It doesn’t work well on a drug test.
That’s why the Sub Solution is such a superstar among synthetic urines. The Clear Choice, company that makes it, had nailed down the right combination of urea, uric acid and creatinine from the start.
They update the formulation every year to make sure it will beat a drug test every time. Double-checking every parameter – from specific gravity to pH and chemical levels that occur in human urine naturally – really pays off.
The Sub Solution has been voted the best and most reliable synthetic urine not only in 2022 but in previous years – 2020, 2019, and 2018 – as well.
The idea so frequently updating the formulation is to ensure the labs can’t even come close to telling the difference between the Sub Solution and normal clean urine.
With cheaper fake pee kits, you will have a ‘good batch’ vs ‘bad batch’ situation. Get a good batch, you’re fine. Get a bad batch, and you’re in the world of hurt.
Thus far the Sub Solution is the only synthetic urine on the market that doesn’t know the meaning of ‘bad batch’. This synthetic urine has a 17-year long proven track record. That alone is more than enough proof of its reliability.
In 2022, Clear Choice introduced their new packaging of the Sub Solution – the shinning green one:

One of the most important aspects of using synthetic urine is to raise the temperature to body temperature. The Sub Solution has a patented heat activator with Lithium Chloride that raises the temperature up to 94°F to 100°F.
Time needed: 5 minutes.
Here is the step-by-step instructions of how to use the Sub Solution. It takes less than 5 minutes:
- Fill the plastic contained with lukewarm tap water.
The water level should be just below the cap.
- Add the contents of the synthetic urine vial to the container and mix gently.
All the ingredients of the synthetic urine are in powdered form. Mixing them with water will create a solution that looks, feels, smells and even bubbles like real urine. (You should mix the whole thing up to 8 hours before use)
- Heat the mixture to 94°F to 100°F. With heat activator (optional) or by keeping it close to your body.
The temperature strip on the plastic container should read anywhere between 94°F and 100°F. You can add a heat activator that heats up the mixture (you will need only about 1/3 of the whole heat activator vial).
- Use it within 8 hours.
Make sure you mix the Sub Solution an hour or so before use. If the solution is mixed more than 8 hours before use, it will go stale (like real urine might go stale and smelly).
- (Optional) Get the practice kit to practice heating up and pouring from the plastic container.
If you’ve used Sub Solution before, you pretty much have it figured out. For people who will be using the Sub Solution for the first time, the practice with practice kit will make you feel much more confident during the real thing. You can get the package of Sub Solution + Practice kit here.


The flipside of having the most reliable synthetic urine is the price. Some of the ‘good batch, bad batch’ cheap fake pee kit might cost $40 or even $30. The Sub Solution, on the other hand, has an $80 price tag.
The reliability when trying to pass a drug test can pay off tenfold, obviously. Not getting or even losing a job is a financial knock-out punch. Nobody really wants to take chances here – and that’s why the price of the synthetic urine itself doesn’t matter as much as the reliability.
You can get the Sub Solution, the most reliable synthetic urine in 2022, at the official TestNegative store here:
#2 Quick Luck: New Premixed Fake Pee Kit
Quick Luck is new synthetic urine. It was launched in 2019 by Clear Choice. It’s basically the same formula as the Sub Solution with one difference: it comes premixed.
That saves you the hassle of 1 or 2 steps when you want to prepare the synthetic urine solution for use.
You also get some heat pads to warm up the Quick Luck or keep it warm. The temperature strap on the plastic container with premixed fake pee will tell you when you’re between 94°F and 100°F. That’s the sweet spot and it’s imperative for you to deliver the urine sample within this temperature interval.
Having a premixed synthetic urine with a superb formula makes all the handling so much easier:
The way to use the Quick Luck is pretty much the same as the instructions on how to use the Sub Solution. All that without needing to mix it yourself. You can check the instructions in the Quick Luck review here.
The best thing about the premixed synthetic urine is that you don’t have to add water, shake everything, and follow those additional steps. More step means more can go wrong.
Because it’s easier to use and has two extra heat pads, the price of the Quick Luck is higher – $100. Hundred bucks vs. losing a job can’t really compare, right?
You can get the Quick Luck at the official store here:
#3 Quick Fix: Most Popular Cheap Kit
The Quick Fix made by Spectrum Labs has almost a legendary status by now. In fact, when most people think about synthetic urine, they think about Quick Fix.
The reason is quite simple: the Quick Fix is the best-selling synthetic urine because it’s much cheaper than the Sub Solution. It costs about $40; however, that doesn’t mean much if the lower price comes at a cost of reliability.
The cheap synthetic urine kits have that ‘good batch, bad batch’ history. Quick Fix is a rare occassion of a cheap synthetic urine that is quite reliable.
However, there was a well-known incident with one of their batches in 2016. Apparently, due to lower-than-needed creatinine levels, the whole batch was bad and a lot of people got caught using the Quick Fix fake pee. You can read more about that in our Quick Fix review.
Obviously, people are afraid such a batch will happen again. They have introduced the new formula to eliviate some concerns:

The Quick Fix is premixed. That means you’ll have less hassle of preparing the synthetic urine mixture. On the other hand, however, the expiry rate is reduced.
The popularity of Quick Fix was somewhat diminished with that ‘bad batch’ incident. Hopefully, the new formula (complete with the new packaging) will have a better track record.
The Quick Fix is the only cheap urine with quite a good, but not intact, reputation. You can get it here:
#4 Powdered Urine Powder by TestClear (Just Add Water)
You know how they make 100% orange juice? In fact, it’s not directly squizzed out of the oranges. Instead, orange juice is squeezed out at the orange farm, the juice is processed to remove water and you get a powdered form of orange juice. Just add water and you will get pristine orange juice.
TestClear used the same logic with their version of synthetic urine. Well, it’s not even synthetic urine; it’s something even more authentic. It’s actual human urine with the water removed.
You have to mix the powdered urine yourself to create a mixture. For that you don’t get a big and awkward plastic container; you can a slim plastic test tube that is much easier to hide:
You get all the instructions on how to use and some advice with the whole kit. They are also laid out in the TestClear powdered urine kit review here.
Needless to say, the TestClear’s powdered urine has a very good track record and it is available at quite an affordable price (it costs about $50).
You can get it here:
Synthetic Urine FAQ
Fake pee has a number of different uses. Synthetic urine can be used for all kinds of practical jokes, as an animal repellent (Mosquitos apparently don’t like the smell of urine), “Golden showers” (Yes, people who are into urination like some extra urine), and for passing a urine drug test (In fact, this is the No. 1 use of synthetic urine nowadays).
More than 90% of all sales of synthetic piss account for people using to pass a drug test. It’s not difficult to see why, given that somebody’s life can be ruined by testing positive for drugs.
Why is it so popular?
Well, it was designed to calibrate laboratory equipment that is used for testing urine. If it is prepared and used correctly, there is no way any lab will detect it as synthetic urine. Lab tests simply detect it as clean real urine.
In essence, it is a complex chemical solution that mimics real urine so well that not even a laboratory drug panel kits can tell a difference between synthetic and real urine.
To answer what synthetic urine is we need to answer what real urine is.
Urine is a complex chemical solution; a by-product of our metabolism. An average person will eliminate about 1.4 liters of urine per day. It contains several different substances such as urea, uric acid, creatinine, has a certain pH and specific gravity, and so on.
The very same can be said for synthetic urine. It is designed to reflect real urine perfectly in every way.
In the end, there is no difference between what comes out of our urinary canal and high-quality synthetic urine.
Real urine is a yellowish liquid that is made out of water, organic and inorganic matter. What is in the synthetic urine? Exactly the same chemical substances that are in real urine.
Important: the contents of specific ingredients of synthetic urine have to be just right to perfectly mimic real urine. The difference between the best synthetic urine and the worst is often how on point the quantity of urinary substances is.
Here is an example of the list of the ingredients that synthetic urine is made out of:
Urea – 50 to 100 mg/dl.
Uric acid – 5.0 to 7.0 mg/dl.
Creatinine – 35 to 320 mg/dl.
Potassium sulfate (K2SO4) – 1 to 3 g/l.
Sodium sulfate (Na2SO4) – 1 to 3 g/l.
Ammonium phosphate – 0.5 to 1.5 g/l.
Ammonium diphospate – 0.5 to 1.5 g/l.
Calcium chloride (CaCl2) – 0.1 to 0.5 g/l.
Magnesium chloride (MgCl2) – 0.25 to 1 g/l.
Why exactly these ingredients?
Urine tests measure 3 specific substances – urea, uric acid, and creatinine.
They are present in every normal urine; in fact, we know that there is at least 50 mg/dl of urea in normal urine. However, the levels of urea have to be below 100 mg/dl.
This is precisely the range of uric acid that every synthetic urine that can be used to drug test has to match. Most of them do; nonetheless, that doesn’t mean that some don’t. Especially looking at the cheaper brand here.
The sulfate, phosphate and chloride salts are included to give synthetic urine the same pH and specific gravity. These are the two most important figures fake urine has to match – pH between 4 and 8 and a specific gravity between 1,000 and 1,035.
If the ingredients of synthetic urine fail to deliver these two metrics, the lab technician analyzing your urine will know something is up.
Fortunately or unfortunately, it does. Literally, thousands of people pass a urine drug test by using synthetic urine kits.
Laboratories that test for drugs are pretty much found helpless to prevent the surge of high-grade synthetic urine used for drug testing. Some states even went so far as to make the sales of synthetic urine illegal because that’s the only way to prevent its use on drug tests.
Most of the people who pass drug tests with flying colors are very confident to recommend working synthetic urine to everybody in need of it.
While it is true that some brands in the world of synthetic urine kits are better than others, it is wise to be aware of problems some cheap fake urines might have.
With high-quality brands, there are no problems because all the ingredients are just right. Some even take care of how to heat up the synthetic urine to body temperature.
A lab cannot test for synthetic urine directly.
However, they can become suspicious if any of the metrics are a bit off.
Here are the four most common reasons people are caught due to using bad synthetic urine:
The high specific gravity of fake urine. Some synthetic urine producers might sulfate and chloride salt addition. This, in turn, raises specific gravity over 1,035 and can be seen as a consequence of using low-quality synthetic urine.
Low-end creatinine. Usually, there are two explanations for creatinine levels under 35 mg/dl. Either there is some kind of kidney problem or the test subject is using a 30$ synthetic urine kit. Good synthetic urine should have at least 100 mg/dl creatinine levels.
Uric acid content is out of range. The normal interval of uric acid concentration is rather narrow (5.0-7.0 mg/dl). The chemical makeup of fake pee has to be just right here; better the quality of the urine, less likely urine will test like synthetic urine.
The temperature of fake piss. Cold urine won’t do because lab tech will know immediately that it didn’t come out of your urinary system. Synthetic urine needs to be heated up to body temperature using heat activators, heat pads, or if you feel like playing with your life, heating it up in a microwave for 10 seconds.
90 to 100F is good.
98 to 102F is perfect.
Temperature, of course, is vital. We can buy the best synthetic urine with perfect urea and creatinine levels but every fake pee also has to be heated to the body temperature.
Urine for drug tests must not be too hot or too cold. Real urine’s temperature is never above 105F (unless you have a really high fever). It also shouldn’t be below 90F. It should be just right – as if it came out of your body minutes ago.
There are a number of ways how to heat up synthetic urine:
Keep it close to your body. This could be very convenient if it worked. The reality is that it’s very difficult to heat a full 5-ounce contained to 100F with just the heat created by our groin or armpit. Not really recommended.
Use heat pads. Several synthetic urine suppliers also offer head pads (they’re like 5$ at most). Some people had good experience using such pads to heat up liquids, others say its really hard to do it and to know that’s exactly 100F as it should be.
Heat activator. The best synthetic urine kits already come equipped with heat activators. Usually, you get granules in a separate vial, put them into the urine and when it heats up to the body temperature, the indicator turns green.
Using the microwave for 10 seconds. That might actually work but it is quite a risky business. How do you know the temperature is not 80F or 120F? One second can make a big difference.
When the fake piss is enough hot, test subjects usually just strap it to a leg or arm in order to keep the heat during the whole process of taking a urine drug test.
Many users keep synthetic urine stocked at home for when the time comes. It makes a lot of sense to have it ready – but you have to ask yourself one question: Does synthetic urine go bad?
As with every product, even synthetic urine has a shelf-life after which it will be unusable (or at least might taste weird – for “golden shower” guys).
After what time does synthetic urine go bad?
Usually 2-3 years.
Of course, it depends on the specific product, batch, and so forth. Most of the kits have the product expiration label on the packaging.
If you are getting tested for drugs on a regular basis, it makes sense to stock up on it in order to be ready when another testing comes around.
Some companies may test their employees every 3 months or so. Having 4 synthetic urine kits at home will last for 1 year and the fake pee usually won’t go bad in 2 years or so.
When buying synthetic urine online, ask for the expiration date. If it’s less than a year, it doesn’t make sense to stock up on synthetic urine near the expiry date.
Keeping urine in the fridge might prevent it from going bad for a longer time. Just keep it out of the freezer (it will turn into ice and likely explode).
You can purchase synthetic piss in two places:
Online (obviously).
In stores near you.
If you’re in a hurry (as in ‘I have a drug test in 5 hours’), it’s best to find a head shop near you that sells it.
If you have a drug test in 2 days or more, it’s better to order it online. At least that’s what most people who wish to pass a drug test do.
Why?
Discreteness and express delivery.
If you buy synthetic urine in a store, you have to physically go to the store and put it on the counter. Everybody knows you’re probably not using it for “golden showers” or as an animal repellent, and most of the stores have cameras and video surveillance.
Online stores that stock synthetic urine are very serious about privacy. They also offer quick delivery and some deal offers.
When in need of passing a drug test, it’s better to go for high-quality urine than for a cheap one. Much can be on the line and users don’t really want to risk it with a questionable 30$ fake pee.
How much does synthetic urine cost?
30$ to 100$.
Most people are much more comfortable spending more on synthetic urine to guarantee they will pass the test. Price is quality in this case.
Conclusion And Advice
Using fake piss has become standard practice when in need of passing a drug test. More and more people this way in order not to lose a job or promotion.
However, a smart way of guaranteeing you will be safe is just not using illicit drugs. The world is becoming more and more accepting of drugs like marijuana and it is for all of us to bring forth progress as far as sensible legislation is concerned.
Scientific researches and other sources:
(1) Goodwin RS, Darwin WD, Chiang CN, Shih M, Li S-H, Huestis MA. Urinary Elimination of 11-Nor-9-carboxy- 9-tetrahydrocannnabinol in Cannabis Users During Continuously Monitored Abstinence. Journal of analytical toxicology. 2008;32(8):562-569.
(2) Hadland SE, Levy S. OBJECTIVE TESTING – URINE AND OTHER DRUG TESTS. Child and adolescent psychiatric clinics of North America. 2016;25(3):549-565. doi:10.1016/j.chc.2016.02.005.
(3) Lee C, Vo TT, Cohen AS, et al. PROFILES OF URINE DRUG TEST IN CLINICAL PAIN PATIENTS VERSUS PAIN RESEARCH STUDY SUBJECTS. Pain medicine (Malden, Mass). 2016;17(4):636-643. doi:10.1111/pme.12900.
(4) Barr DB, Wilder LC, Caudill SP, Gonzalez AJ, Needham LL, Pirkle JL. Urinary Creatinine Concentrations in the U.S. Population: Implications for Urinary Biologic Monitoring Measurements. Environmental Health Perspectives. 2005;113(2):192-200. doi:10.1289/ehp.7337.